
High-performance cell atlas workflow driven by manifold fitting
February 26, 2026Researchers from the National University of Singapore (NUS) have developed CellScope, a high-performance single-cell analysis framework that uses manifold fitting to analyse single-cell RNA sequencing (scRNA-seq) data. This framework helps build detailed “cell atlases” that map different cell types and show how they further group into finer subtypes.
As single-cell atlases grow in size and complexity, many existing tools struggle to separate meaningful biological signals from noise and fail to capture the dynamic and hierarchical nature of cellular organisation. They often rely on single-level gene markers and limited clustering resolution, making it difficult to explore how broad cell types branch into specialised subpopulations.
The research team led by Associate Professor YAO Zhigang from the NUS Department of Statistics and Data Science developed CellScope, a framework for constructing high-resolution “cell atlases” at multiple clustering levels. CellScope is based on the idea that, although each cell is measured across thousands of genes, the meaningful biological difference between cells can often be described in a lower dimensional structure (a “manifold”). However, these measurements are affected by two major sources of noise: housekeeping genes that are active in almost all cells and do not help distinguish cell types, and technical noise from the sequencing process. Using a two-stage approach, CellScope first performs gene selection to distinguish the signal from housekeeping-driven variation, then denoises cell representations to improve separation of closely related populations. This work was carried out in collaboration with Professor Jessica LI from the Fred Hutchinson Cancer Center at University of Washington.
The research breakthrough was published in the journal Nature Communications.
The framework integrates several key components in one workflow. This includes adaptive gene selection, denoising based on manifold structure, hierarchical clustering across multiple resolutions, and a tree-style visualisation that shows how clusters relate to each other. It also introduces a dynamic “molecular identity” system that tracks how the importance of genes can change at different clustering levels, rather than labelling genes as simply “marker” or “not marker”.
CellScope was evaluated on 36 single-cell datasets from human and murine model tissues. When compared with widely used tools, CellScope more often produced results that matched known cell labels, and it did so without requiring extensive manual fine-tuning. Beyond these performance tests, CellScope also helped researchers uncover new biological insights. For example, it identified immune cell changes in COVID-19 patient blood samples that became more pronounced with increasing disease severity.
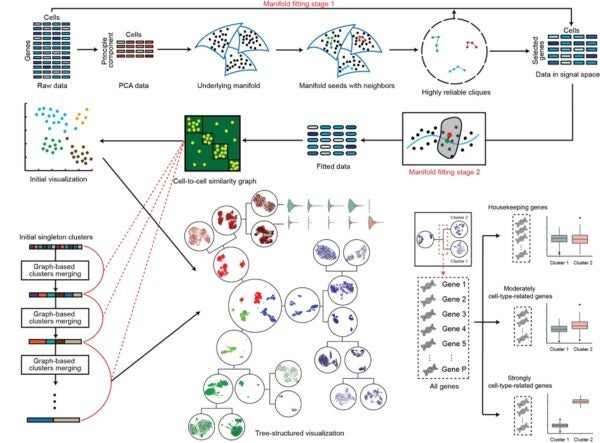
Figure shows an overview of the CellScope workflow. CellScope first selects informative genes and reduces noise using a two-step manifold fitting process, then builds a cell-to-cell similarity graph to cluster cells into a hierarchy of cell types and subtypes. It outputs a uniform manifold approximation and projection (UMAP) map of the cells and a tree-style view showing how clusters split, along with genes grouped as housekeeping, moderately cell-type-related, or strongly cell-type-related.
Prof Yao said, “CellScope represents more than an incremental technical improvement. Rather, it is a fundamentally new framework built upon the theoretical foundation of manifold fitting.”
“By bringing rigorous geometric ideas into single-cell analysis, CellScope introduces an innovative way to estimate the intrinsic low-dimensional structure underlying complex biological data. At the same time, it enables the framework to achieve high accuracy, computational efficiency and strong interpretability without extensive manual adjustment,” added Prof Yao.
Moving forward, the team plans to continue developing and maintaining CellScope, including expanding its compatibility with emerging data types such as spatial transcriptomics and multimodal omics, and enabling systematic reanalysis of large public datasets to uncover additional cellular subtypes and disease-associated signals.
Reference
Li B; Lin R; Ni T; Yan G; Burns M; Li JJ*; Yao Z*, “CellScope: high-performance cell atlas workflow with tree-structured representation” Nature Communications DOI: 10.1038/s41467-025-67890-3 Published: 2025.
